Maladies rares :
Syndrome de Marfan
C'est une maladie héréditaire du tissu conjonctif, correspondant à une anomalie d'une protéine de structure appelée fibrilline.

Cliquez pour agrandir l’image
C’est une maladie héréditaire du tissu conjonctif, correspondant à une anomalie d’une protéine de structure appelée fibrilline. Par une meilleure connaissance de celle-ci, on diagnostique maintenant plus précocement des formes frustres et incomplètes du syndrome de Marfan.
La maladie touche autant les garçons que les filles. On estime à 5000 le nombre de cas en France. La fréquence de la maladie est d’environ 1 pour 10 000 naissances.
Manifestations cliniques
Les symptômes, pas toujours présents chez chacun des patients (polymorphisme important) sont constitués, principalement et de façon caractéristique, de modifications squelettiques , oculaires et cardio-vasculaires.
Modifications squelettiques
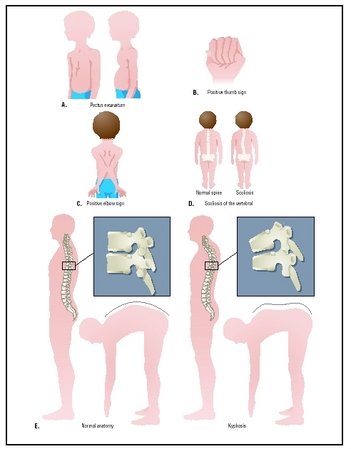
Grande taille avec membres anormalement longs, doigts et orteils longs et fins (arachnodactylie ou dolichosténomélie), hyperlaxité ligamentaire, déformations thoraciques (dépression, protrusion), cyphoscoliose, palais ogival.
Modifications oculaires
Subluxation du cristallin, habituellement vers le haut (50 % des cas) avec parfois décollement de la rétine, glaucome ou autre anomalie du cristallin.
Modifications cardio-vasculaires
Les plus graves : fréquents prolapsus de la valve mitrale et dilatation de l’aorte, avec risque de dissection et rupture d’anévrisme (dans environ 10 % des cas, mort subite).
D’autres malformations cardiaques (surtout communication inter-auriculaire) ou pulmonaires (emphysème, pneumothorax) peuvent lui être associées.
Diagnostic
Dans les formes complètes le diagnostic est aisé.
Toutefois l’aspect marfanoïde typique, les dislocations des cristallins et les anomalies cardio-vasculaires peuvent chacun être transmis indépendamment dans certaines familles.
Le diagnostic n’est donc habituellement pas fait à moins qu’un des membres de la famille au moins, n’ait des modifications caractéristiques concernant au moins deux des trois systèmes de tissu conjonctif.
Le diagnostic est des plus facile à établir quand le malade ou des membres de sa famille ont des signes objectifs de subluxation cristalline, de dilatation aortique, et une cyphoscoliose sévère ou des déformations thoraciques.
Il faut faire un examen à la lampe à fente et un électrocardiogramme pour tous les malades chez lesquels le diagnostic est suspecté.
Comme les gènes responsables ont été identifiés, un test moléculaire est disponible pour confirmer le diagnostic; la consultation d’un généticien le permet aussi.
Deux signes cliniques évocateurs et faciles à constater: le signe du pouce (lorsque le pouce est à l’intérieur du poing fermé, son extrémité dépasse) et le signe du poignet (lorsqu’une main entoure le poignet de l’autre bras, le pouce et le petit doigt se croisent).
Transmission
La maladie est transmise sur le mode autosomique dominant, à pénétrance variable.
15 à 30 % des cas peuvent être dus à de nouvelles mutations spontanées.
Le “saut de génération” lié à une expressivité variable est relativement commun.
Un des gènes en cause se situe sur le chromosome 15, un autre sur le chromosome 3.
Pour plus d’informations :
Association Marfans